
Moving Toward the Implementation Deadline – Approaching MDR Compliance

As we move toward the implementation of the Medical Device Regulation 2017/745/EC (MDR), industry reports indicate many companies still seem unprepared as they struggle to understand its requirements.1 In this blog post, PPD’s Michelle O’Connor, senior director of regulatory affairs, Kenneth Butz, director of regulatory affairs, and Johan Brandt, principal regulatory affairs specialist, discuss the MDR and provide suggestions to device manufacturers to support compliance.
A recent survey by Greenlight Guru found that only 53% of companies were likely to be ready in time for the implementation of the MDR, due primarily to resourcing issues.2 Regulatory challenges were reported as the most significant challenge by >70% of responders in a recent Emergo survey.3 An article from 2017 still remains valid today in that there is still a degree of confusion in the industry regarding MDR implementation and steps to be taken to meet its requirements.4 As a first point, device manufacturers should be aware they can avail themselves of a three-year transition period (per Art. 120 of MDR) to renew their Medical Device Directive 93/42/EC (MDD) certificates for Class II and III devices by 26 May 2024.5 Next, we address regulatory concerns raised in industry surveys and our recommendations for meeting MDR requirements.
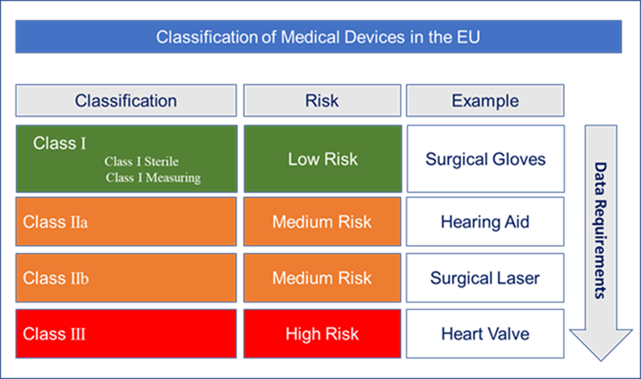
What is changing?
With the overall aim of improving safety and performance, the MDR has wide-ranging impacts on technical file documentation, clinical evaluation, device classification and post-market surveillance. Some key challenges and considerations for transitioning to MDR are outlined below.
- More stringent requirements for clinical evidence, particularly for Class III devices, and increased scrutiny on clinical data based on literature review
Clinical evidence requirements are proportionate to the risk associated with a given device. Per the MDR, a clinical evaluation can be either a critical evaluation of relevant scientific literature, a critical evaluation of the results of all clinical investigations or a combination of both.
With increased focus on robust clinical data supporting device re-certification, including requests by regulators for new clinical data for devices previously certified under the MDD, companies must plan and design clinical evaluations to remediate gaps. Literature reviews may no longer be considered sufficient depending on the class of the device and the intended uses.
- Reclassification of devices
With a risk-based approach to device classification, some devices have been up-classified under the MDR, now placing additional data burden on manufacturers to potentially generate new supporting chemical/biological and clinical data.
Up-classification is based on devices meeting the criteria of Annex IX, Rule 14 classification (refer to page 144 of the MDR). This was previously Rule 13 under the MDD wherein an ancillary substance is effectively a device containing a medicinal substance. For example, under Rule 14, some devices previously classified as Class II devices under the MDD, are now re-classified as Class III devices under the MDR if they contain an ancillary substance. This re-classification generally requires an updated Clinical Evaluation Report (CER) but also, more significantly, the development of Chemistry, Manufacturing and Controls (CMC) information, similar to a Module 3 dossier that is compiled for pharmaceutical products.
- Implementation of Unique Device Identifiers (UDIs) and the establishment of the new European Databank (EUDAMED) for medical device information
This requires products to be correctly labelled with specified safety information and symbols and to meet requirements for electronic “Instructions for Use.” This is an unambiguous identification code specific to the manufacturer and device, which will be listed in the EUDAMED for devices used to track EU-marketed devices, aiding in the monitoring of post-market safety and in the combat against counterfeit devices.
- Rigorous post-marketing surveillance
More emphasis on surveillance and vigilance is required once a device is CE (Conformité Européenne) – marked. A dedicated post-market surveillance (PMS) system based on a PMS plan aligns the monitoring device of performance for recertification, annual safety updates for higher-risk class devices and rapid reporting of safety incidents.
This information will be publicly available, and companies are required to trend and report periodically using Periodic Safety Update Reports.
- Establishment of the “Person Responsible for Regulatory Compliance” (PRRC) role
The manufacturer is required to have at least one person responsible for oversight of regulatory compliance. This PRRC must have a formal qualification in a relevant scientific discipline and experience in regulatory affairs or quality management systems compliance. The PRRC role can be subcontracted depending on the size of the organization.
- The change in the role of the Notified Bodies (NB)
The number of newly designated NBs under MDR is slowly increasing. However, the overall number is significantly smaller than under the MDD, which is placing additional constraints on the entire sector in relation to the conduct of conformity assessments. Manufacturers are vying for position with the limited number of NBs supporting both re-certification under the MDD and new certifications under the MDR. Many NBs have ceased operations due directly to the new requirements under the MDR.
- Managing the nuances of Brexit
European Union notified bodies located in the U.K. lost their EU accreditation because of Brexit. Additionally, authorized representatives (AR) in the U.K. are no longer authorized for the EU market, and importers located in the U.K. can no longer import products for the EU market. However, MDR will apply in Northern Ireland, which now requires that U.K. manufacturers will need an EU or Northern Ireland-based AR. However, some products for the Northern Ireland market will still require registration with the MHRA. The U.K. will continue to recognize the CE mark and certificates issued by EU-based NBs until 30 June 2023, although a new route to market and product marking (U.K. Conformity Assessment [UKCA]) became available for manufacturers intending to place a device on the U.K. market beginning on 1 January 2021. Starting on 1 July 2023, it will be mandatory for device manufacturers intending to place a device on the U.K. market to have the UKCA mark.
The path forward – best practices
Any company intending to market new or legacy products must now comply with the MDR to obtain a CE mark. Manufacturers must upgrade their processes and technical documentation, in accordance with the new regulation. Strategies for success include:
- Review the MDR classification rules to determine whether new conformity assessment routes now apply.
- Perform a gap analysis of all processes and device technical documentation, and prepare a comprehensive plan for compliance with the more prescriptive MDR safety and performance requirements.
- Work collaboratively with an NB to ensure MDR compliance including strategy and schedule.
- Prepare for more stringent clinical evidence requirements by reviewing CERs for compliance with MEDDEV 2.7.1 (Revision 4) and MDR, and define a robust process and strategy with a cross-functional team approach for incorporating relevant information and updates.6
- Review systems and procedures for more stringent vigilance and post-market surveillance data requirements based on the PMS plan and supportive post-market clinical follow-up plan.
- Align processes for end-to-end product traceability that support both the UDI system and the central EUDAMED database.
Conclusions
Many of the changes brought on by MDR, including increased pre- and post-market clinical data requirements, affect multiple functions within an organization and also their business partners and NBs. With the focus of the MDR on the entire product life cycle, a comprehensive and holistic approach is required.