
The Present and Future of Conditional Regulatory Authorization and Access Pathways for ATMPs
Advanced Therapy Medicinal Products (ATMPs), such as cell therapies, gene therapies and tissue-engineered therapies, represent some of the most technologically innovative treatments today. Since ATMPs can dramatically improve patient outcomes, demand for market access is on the rise, requiring the exploration of a detailed commercial approach.

Charting the current landscape for cell and gene therapies and ATMP development
In surveying the market, advanced therapy medicinal products (ATMPs) — including cell therapies, gene therapies and tissue-engineered treatment — represent one of the more promising therapeutic development lines. Taken as a group, these technologically innovative treatments potentially offer differentiated — often novel — clinical benefits that can dramatically improve patient trajectories across a host of underserved indications, like cancer.1 The sophistication of these interventions, from their manufacturing process and administrative complexity to their impact on disease courses and complex treatments, directly contrasts with standard therapeutics. Additionally, market access considerations for these products — from what constitutes reasonable pricing to how an ATMP’s clinical value is determined — requires a nuanced commercialization approach.
After years of iterative development, manufacturers, clinicians and patients remain cautiously optimistic that these products will be favorably evaluated by regulatory overview and health technology assessment (HTA) gatekeepers. While over 20 ATMPs have obtained national regulatory approval or marketing authorizations in one or more global markets, the industry continues to face the nascent stages of addressing market access for these technologies, as the pipeline of prospective and investigational ATMPs significantly outpaces commercialized treatments. Whether seeking approval, coverage and reimbursement in the U.S., where the Food and Drug Administration distributes ATMPs in two broad categories (Figure 1), or unlocking access in Europe, with the European Medicines Agency assigning twice as many classifications (Figure 2), understanding how to maximize market access opportunities as part of broader strategies is key.


Understanding the “SUC” driving successful ATMP market access
Many ATMPs are genuinely unique interventions, from modified autologous stem cell treatments to first-in-class biologic therapies for cancers and rare diseases. Differentiated as they may be, ATMPs all share three characteristics that substantiate some of their value to regulatory and HTA stakeholders.
- Scarcity: This concept refers to both the lack of existing treatment options for indications addressed by ATMP development and the comparably small patient populations associated with these indications. The value of any effective intervention increases in situations where therapeutic scarcity is observed.
- Urgency: In addition to lacking effective interventions, many indications targeted by ATMPs are also severe disorders that have poor prognostic trajectories. Without the benefits of an effective therapy, patients face acute quality of life declines or increased mortality rates, making streamlined ATMP access vital.
- Complexity: Development and administration of ATMPs can be resource-intensive processes, without always having accompanying patient population volume as an offset. Accordingly, attempts to recoup costs are reflected, to some degree, in proposed product pricing, alongside these therapies’ health economic value.
Combined, these features foster a need for manufacturers to use dynamic, strategic market access approaches to ensure desired outcomes and maximum realized value when interacting with regulatory and HTA stakeholders.
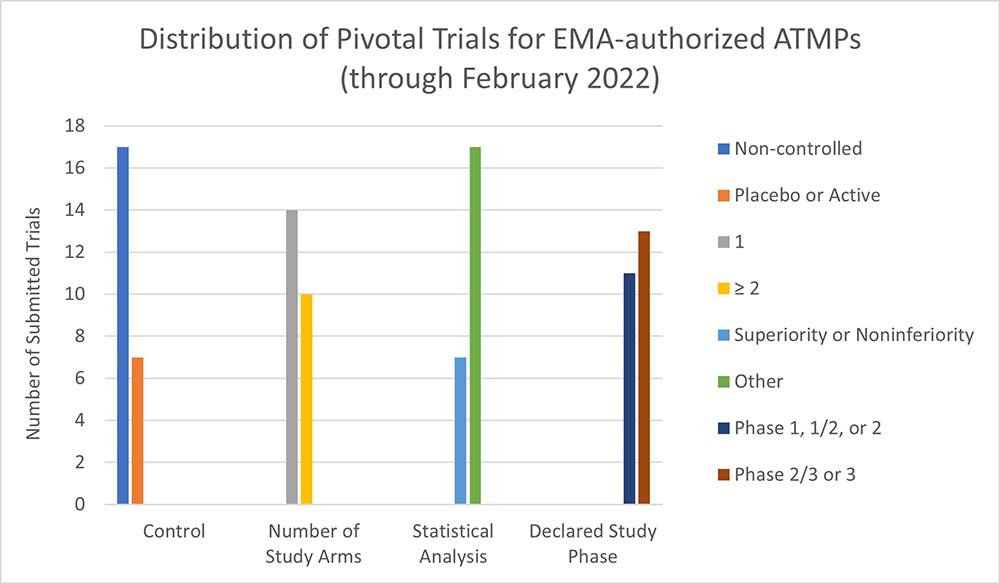
These characteristics can lead to an increased likelihood that an ATMP is presented to market access stakeholders with relatively incomplete clinical evidence packages. Similarly, common challenges in executing clinical trials for ATMPs (e.g., study size, selection [or absence] of comparator, lack of long-term outcomes data) often lead to these products having insufficient materials to satisfy HTA stakeholder requirements, particularly in markets where cost-effectiveness and durable efficacy are needed to inform evaluations. For instance, recent analysis (e.g, Iglesias-Lopez et al., 2021; Evidera analysis) finds that the majority of ATMPs submitted for regulatory approval in Canada, the U.S. and Europe are supported by relatively less-rigorous evidence, with a preponderance of pivotal non-randomized controlled trials, compared to approved non-ATMPs submitted during the same period.2
Most regions offer regulatory approval options intended to support ATMPs and other innovative therapies prior to manufacturers obtaining evidence dossiers with fully developed clinical data, patient-reported outcomes and health-economic data. The provisional mechanisms and pathways — known by a variety of names — can facilitate commercialization and sustained development of ATMPs, when used effectively. The European Medicines Agency, among others, favorably frames the purpose of these pathways toward pioneering ATMPs, with eligibility for PRIME (Priority Medicines) and other accelerated pathways contingent on many of the same factors (e.g., the “SUC” construct above) inherent to cell and gene therapies.3 Given this positive environment, the top considerations for manufacturers shift to the following three concerns.
- How can ATMP manufacturers make their assets eligible for one or more accelerated access pathways?
- What strategies and steps can be leveraged to mitigate risk for clinical trial development and market access submissions?
- Are any efficiencies available between optimizing a therapy for regulatory stakeholders and framing the same treatment for HTAs?
Where next for advanced therapies?
As market access programs for ATMPs evolve in various markets, it is increasingly important for manufacturers to incorporate proactive tactics to secure desired commercial results. It’s expected that regulators will continue to heavily employ conditional authorization channels, necessitating corresponding tactical shifts by innovators. In short, recommended best practices boil down to being “forward and focused.” Future ATMP market access success will be built on deliberate advance actions, with the aim of creating a compelling, targeted and evidence-driven value proposition for an asset. Three key activities can bolster this aim.
Early scientific advice
Market access stakeholders are constantly iterating policies, statutes and perspectives regarding evaluation and assessment of new technologies. Consequently, this magnifies the impact of engaging these groups earlier in the development cycle to align on recommendations for comparators, study design and size, endpoints, and study enrollment criteria. Formal interaction programs and protocols are codified for several traditionally high-priority markets, and manufacturers are encouraged to leverage internal staff or expert external collaborators to engage regulatory or joint regulatory-HTA scientific advice. Recent regulatory guidance and independent studies show that, when leveraged appropriately, early scientific advice resources yield tangible benefits, including comparably higher authorization/approval rates and shorter evaluation times for a range of therapeutics, vaccines and diagnostics.4,5,6
Deliberate, clinically motivated product profiling and positioning
An ATMP’s regulator- and payer-resonant placement in a treatment algorithm, or its ideal addressable patient population, are among the most important insights that can be elicited from early scientific advice and should be addressed even if that engagement opportunity is not exercised. Manufacturers are well served to solidify these two factors and adhere to them across the development cycle, as the coherence of the cumulative narrative — clinical study data, monographs and other materials included — plays a critical role in stakeholder perception. Issues such as misalignment between a product’s studied population and its proposed label frequently manifest in negative or substantially delayed positive recommendations from HTA stakeholders, even with favorable, disrupting commercialization programs. A dedicated, multifaceted approach to communicating product profiles, reinforced with consistent alignment in clinical trials, can effectively mitigate some of these potential complications.
Long-term strategic evidence generation
Investing in preclinical-to-commercialized evidence planning is the final and most holistic piece of a comprehensive market access strategy. Rather than simply seeking to develop robust, gold-standard trials to support marketing authorization, ATMP manufacturers are better served by drafting plans to generate robust evidence that reinforces
- desired regulatory approval (efficacy and safety),
- health economic value (durable benefit and cost-effectiveness),
- market differentiation (defined clinical use case), and
- maturation (establishment as or among standards of care).
This sustained chain of evidence generation across product life cycles can help realize initial access gains and reduce the likelihood that subsequent stakeholders force additional outlays — new studies, resubmissions, etc. — rather than accept value demonstrated by pre-designed extension trials, registries and real-world evidence studies.
While the prospects for ATMPs are promising for pharmaceutical developers, clinicians and patients, thoughtful development and planning are essential to ensure that the full value of these therapies can be realized by all parties.